Lessons Learned

Welcome to Lessons Learned!
We have compiled questions that might arise in the course of your clinical research project.
We hope that you find these helpful.
Schreiben des Studienprotokolls – eine kleine Auswahl an Dos und Don’ts
Schreiben des Studienprotokolls – eine kleine Auswahl an Dos und Don’ts
Für viele stellt das Erstellen des Studienprotokolls, insbesondere das Schreiben selbst, eine grosse Herausforderung dar. Bei der Beratung und beim Review von Studienprotokollen stellen wir in vielen Fällen immer die gleichen, wiederkehrenden Probleme fest. Dieser Artikel soll in verkürzter Form eine Auswahl der häufigsten (technischen) Fehler beim Schreiben von Studienprotokollen wiedergeben, mit Tipps, wie diese vermieden werden können. Es geht hierbei nicht um methodische oder regulatorische Aspekte. Ein Studienprotokoll ist ein komplexes Dokument, bei dessen Erstellung idealerweise eine Vielzahl von Personen beteiligt ist. Software, insbesondere Textverarbeitungs- und Literaturverwal-
tungsprogramme, unterstützt einen bei diesem Prozess. Deshalb ist es essentiell, dass man als Autor:in eines Studienprotokolls Expert:in für die gesamte Funktionalität solcher Software ist (oder wird). Die hier vorgestellte Auswahl erhebt keinen Anspruch auf Vollständigkeit, und die vorgeschlagenen Lösungen sind aus unserer Erfahrung gewachsen und sicher nicht in jedem Einzelfall die beste Variante. Auch sind die vorgeschlagenen Lösungen häufig nicht detailliert genug. Sie dienen vielmehr als Anregung. Technische Details und Anleitungen findet man aber schnell auf Webseiten oder YouTube.
Die Zusammenfassung aus der (Swissethics-) Protokollvorlage wird als
Arbeitsinstrument genutzt
Häufig wird die Projektidee anhand des strukturierten Zusammenfassungskapitels aus der Swissethics-Protokollvorlage entwickelt, das heisst, hier wird die Projektidee erstmals formuliert, adaptiert, Feedback von Kolleg:innen eingeholt usw. Häufig wird dann parallel schon am Haupttext des Protokolls gearbeitet. Dies führt in praktisch allen Fällen zu Inkonsistenzen zwischen Zusammenfassung und dem Haupttext und auch zu Inkonsistenzen im Haupttext selbst.
Tipp: Mit einem separaten Dokument starten, in dem das Projekt anhand des PICO-Schemas (Popula-tion, Intervention/Exposure, Control, Outcome) formuliert wird. Allenfalls dieses weiter ausbauen und Feedback einholen, beispielsweise durch Einfügen weiterer Kapitel (etwa Kapitelüberschriften aus der Zusammenfassungsvorlage kopieren), Hinzufügen des grafischen Study-Flows (angelehnt an das CONSORT-Diagramm) und des tabellarischen Visit-Schedules. Nun die Inhalte in die Protokollvorlage übertragen und weiter ausbauen. Weder an der Zusammenfassung noch an den administrativen Kapiteln, zum Beispiel zum Thema Datenmanagement oder Monitoring, arbeiten, es sei denn, diese sind kritisch für das Projekt. Allenfalls für Kommentare und Korrekturen zirkulieren. Erst wenn der Haupttext nahezu fertig ist, die Zusammenfassung und die administrativen Kapitel schreiben. Das fertige Protokoll immer zur Schlusskorrektur jemand anderem geben.
Unklare Versionierung von Dokumentenentwürfen und der Fassung für das Qualitätsmanagement
Ein Studienprotokoll ist ein kontrolliertes Dokument. Aus diesem Grund werden die endgültigen Fassungen versioniert, zum Bespiel nach einem Schema «Version #.#» oder sogar «Version #.#.#», jeweils mit Datum. Diese Versionen sind zu unterscheiden von den (vielen) Versionen, die beim Erstellen einer Studienprotokollversion anfallen. Wenn kein kollaboratives Tool (z.B. Google Docs, MS Office 365) zum Schreiben genutzt wird, müssen Änderungen und Kommentare durch die Ko-Autor:innen nachvollziehbar bleiben.
Tipp: Der oder die Hauptautor:in ist alleinverantwortlich für das Management der verschiedenen Entwurfsversionen. Entweder zirkuliert man das Dokument sequentiell unter den Mitautor:innen oder zur gleichen Zeit (Zeitverlust versus bessere Übersicht). Mitautor:innen sollten dann ihr Kürzel an den Dateinamen anfügen, zum Beispiel «…_st.docx». Der oder die Hauptautor:in arbeitet alle Vorschläge ein und benennt die Datei mit einer neuen Entwurfsversion, basierend auf einem vorher festgelegten Benennungsschema. Die Versionsnummerierung im Dateinamen sollte nur aus fortlaufenden Nummern bestehen und niemals das Wort «final» enthalten. Die endgültige Version sollte immer als saubere, bearbeitbare Datei abgelegt werden. Beim Starten des Überarbeitungsprozesses können so mittels Änderungsverfolgung («Track Changes»; siehe nächster Punkt) alle Änderungen nachverfolgt werden (siehe auch Tipp zur Versionsübersicht weiter unten).
Änderungsverfolgung «Track Changes» wird nicht genutzt
Die Mitautor:innen machen Änderungen am Dokument ohne dass diese nachverfolgt werden können oder markieren Änderungen manuell, etwa mittels farblicher Hervorhebungen.
Tipp: Der oder die Hauptautor:in sollte vor dem Zirkulieren eines Entwurfs immer die Änderungs-verfolgung («Track Changes») einschalten und sich nicht auf die Mitautor:innen verlassen. Wenn das Dokument wegen zu vieler Änderungen unübersichtlich wird, können bestimmte Markierungen ausgeblendet werden, zum Beispiel Formatierungsänderungen oder Änderungen von bestimmten Personen.
Das Studienprotokoll enthält keine Versionsübersicht
Jedes Studienprotokoll unterliegt im Verlauf eines Projekts Änderungen («Amendments»). Diese sind mittels Änderungsverfolgung («Track Changes») zu dokumentieren. Mit der Dauer wird dies jedoch unübersichtlich und erschwert einen schnellen Überblick über die Veränderungen über die Zeit.
Tipp: Beim Inkraftsetzen der ersten Version schon eine Versionsübersicht (Tabelle) in das Studien-protokoll einfügen. Bei jedem Amendment die (wichtigen) Änderungen in dieser Übersicht beschreiben beziehungsweise zusammenfassen.
Die Studienprotokollvorlage von Swissethics wird blind benutzt
Swissethics bietet Vorlagen für das Schreiben von Studienprotokollen für die verschiedenen Studien-typen an. Diese sind ein wichtiges Hilfsmittel und helfen sowohl beim Schreiben als auch bei der Beurteilung. Je nach Projekt sind diese Vorlagen jedoch nicht detailliert genug, und manche Vorlagen sind technisch nicht auf dem neuesten Stand.
Tipp: Aus unserer Sicht muss man sich nicht sklavisch an die Protokollstruktur halten. Anpassungen, insbesondere das Hinzufügen von Kapiteln, sind für viele Projekte sinnvoll. Beispielsweise enthält die Vorlage für die sonstigen klinischen Versuche keine getrennten Kapitel für die Beschreibung der Studienpopulation, Intervention und der Studienuntersuchungen. Auch fehlt in vielen Vorlagen ein separates Kapitel zur Beschreibung der Governance und der Rolle der verschiedenen Studiengremien. Einige Vorlagen haben auch die Formatvorlagen, insbesondere der Überschriften, nicht sauber implementiert. Dies sollte man immer prüfen und allenfalls anpassen (siehe Tipps zu Überschriften im zweiten Teil, Newsletter vom Februar 2024).
Layout
Es wird nicht genug in das Layout investiert, und die Nützlichkeit eines ansprechenden und übersichtlichen Dokuments wird unterschätzt. Um ein neues Kapitel auf einer neuen Seite zu beginnen, werden oft mehrere Zeilenumbrüche («Carriage Return») eingefügt.
Tipp: Ein sauberes Layout ist nicht nur aus ästhetischer Sicht wichtig, sondern auch für die Leser:innen und das Arbeiten mit dem Dokument. Ein sauberes Layout trägt zur Qualität der Studiendurchführung bei. Daher sollte genug Zeit in ein ordentliches Layout investieren werden. Formatierungen (Schriftgrösse, Schriftschnitt, Farben, Spalten, Abschnitte usw.) sollten sparsam, aber gezielt eingesetzt werden. Die Schriftgrösse des Texts sollte nicht kleiner als 11 Pt. sein, der Zeilenabstand sollte grösser
als 1.0 gewählt werden, mit dezidiertem Absatzabstand (meist 50% der Schriftgrösse), und die Seitenränder mindestens 2.5 cm betragen. Wenn Text auf einer neuen Seite beginnen soll und dies nicht durch den Textfluss erfolgt, sollte dieser Seitenumbruch mithilfe der entsprechenden Funktion eingefügt werden und nicht durch mehrere Zeilenumbrüche.
Hinweis: Jedes Hauptkapitel sollte auf einer neuen Seite beginnen.
Überschriften und Text werden manuell formatiert
Auch heutzutage werden Überschriften und Text gelegentlich noch manuell formatiert, zum Beispiel Schriftgrösse und Schriftschnitt (fett).
Tipp: Überschriften und andere wiederkehrende Textteile mit spezieller Formatierung sollten eine dezidierte Formatierung zugewiesen bekommen. In MS Word geschieht dies über Formatvorlagen/Stile («Styles»). Es ist hierbei wichtig, den Unterschied zwischen Absatz- und Zeichenformatvorlagen zu kennen. Auch Tabellen-/Grafiküberschriften, Fussnoten etc. sollten eigene Formatvorlagen erhalten.
Überschriften werden manuell nummeriert
Da ein Studienprotokoll ein komplexes Dokument ist, gibt es verschachtelte Überschriften. Die Hierarchieebene wird hierbei meist mittels Nummerierung dargestellt. Diese Nummerierung erfolgt häufig noch immer manuell und nicht automatisiert. Zudem spiegelt die Formatierung häufig nicht die Hierarchieebene wider.
Tipp: Die Nummerierung von Überschriften, zumindest in MS Word, ist eine Wissenschaft für sich. Es führt leider kein Weg daran vorbei, sich dieses Wissen mittels YouTube oder detaillierten Beschrei-bungen auf Webseiten anzueignen. Alles andere führt zu noch grösseren Frustrationen und deutlich mehr Zeitaufwand. Als Startpunkt dient der folgende Hinweis: Die jeweilige Formatvorlage muss mit einer dezidiert definierten mehrstufigen Liste («Multilevel Lists») verknüpft werden. Die Formatvorlage für die jeweilige Überschriftenhierarchieebene sollte eben diese Hierarchie nicht nur über die Nummerierung, sondern auch über die Schriftgrösse, Absatzabstand vorher und nachher usw. abbilden.
Das Inhaltsverzeichnis
Gelegentlich sehen wir in Protokollen, dass das Inhaltsverzeichnis manuell erstellt wurde. Änderungen an den Überschriften oder Verschiebungen der Seitenzahlen sind deshalb sehr schwer und nur sehr mühsam nachzuarbeiten.
Tipp: Wenn alle Überschriftenebenen sauber mittels Formatvorlagen definiert wurden, lässt sich das Inhaltsverzeichnis problemlos automatisiert erstellen. Hierbei ist es jederzeit möglich, festzulegen, bis zu welcher Hierarchieebene die Überschriften im Inhaltsverzeichnis erscheinen sollen. Die Aktuali-sierung kann beispielsweise in MS Word durch die Tastenkombinationen Strg+A und dann F9 erfolgen.
Bild- und Tabellenüberschriften
Beschriftungen von Grafiken und Tabellen werden manuell formatiert.
Tipp: Jede Grafik, jedes Bild und jede Tabelle sollte mithilfe der entsprechenden Funktionalität («Caption») beschriftet werden. Dies ermöglicht die Erstellung eines separaten Verzeichnisses für die Grafiken, Bilder und Tabellen, sofern dies gewünscht ist. Es können auch eigenen Typen definiert werden. Zudem werden die Elemente automatisch nummeriert, was äusserst hilfreich ist, wenn sie im Dokument verschoben oder neue Elemente eingefügt werden. Ausserdem ist die Nutzung der Beschriftungsfunktionalität die Voraussetzung für saubere Verknüpfungen.
Verknüpfungen zu Überschriften, Grafiken und Tabellen
Verknüpfungen zu Überschriften, Grafiken, Tabellen und anderen Elementen werden manuell erstellt.
Tipp: Zum Erstellen von Verknüpfungen die entsprechende Funktionalität benutzen. In MS Word findet sich dies unter dem Tab «Einfügen/Insert» und dann «Verknüpfung/Cross-Reference». Dies mag anfangs mühsam und unnötig erscheinen. Wer jedoch wiederholt Elemente im Dokument verschiebt oder neue Elemente einfügt (auch wenn man denkt, dies würde nie passieren – es wird passieren…), wird den Wert dieses Aufwands erkennen und die daraus erzielte Zeitersparnis zu schätzen wissen. Eine Aktuali-sierung der Verknüpfungen kann hier wiederum mittels der Tastenkombination Strg+A und F9 erfolgen.
Hinweis: Für standardisierten und wiederkehrenden Text (z.B. Datumsangaben, Studiengrösse etc.) gibt es prinzipiell zwei Varianten, um Änderungen im gesamten Text nachzuarbeiten:
1) Manuelle Methode: Man markiert die jeweiligen Stellen mit einem eindeutigen Marker z.B. «$$$». Wenn der Text angepasst werden muss, sucht man diesen mittels der entsprechenden Funktionalität (in MS Word: Tastenkombination Strg+F, bzw. Strg+H) und löscht die Marker ganz am Schluss.
2) Automatische Methode: Man nutzt die Lesezeichen-Funktionalität («Bookmark») und fügt diese mittels Verknüpfungen («Cross-Reference») ein.
Literaturverzeichnis
Für die Verwaltung der Referenzen wird entweder die Fussnotenfunktionalität oder die eingebaute Referenzenfunktionalität genutzt, oder Referenzen werden manuell im Text erwähnt und dann das Literaturverzeichnis manuell erstellt.
Tipp: Für ein Studienprotokoll muss zwingend ein dezidiertes Literaturverwaltungsprogramm benutzt werden, und dies von Anfang an! Es gibt eine Vielzahl solcher Programme, wie beispielsweise EndNote und Mendeley, einige davon sind auch kostenfrei, wie Zotero und Mendeley. Ein technischer Hinweis: Die Verknüpfungen erfolgen meist mittels Add-Ins und Field-Codes, ähnlich der Seitenzahlnummerierung.
Rechtschreibung und Grammatik – «The Trail»
Die automatisierte Rechtschreibprüfung wird ausgeschaltet oder ignoriert.
Tipp: Auch wenn die automatische Rechtschreibprüfung nicht perfekt ist, ist sie doch ungemein nützlich. Zwar weiss heutzutage jede:r, dass mit «Trail» eigentlich «Trial» gemeint ist, aber unschön bleibt es trotzdem.
Abkürzungen
Wenn es etwas gibt, das klinisch Forschende lieben, sind es Abkürzungen! Diese sind jedoch oft nicht eindeutig oder nicht allgemein bekannt. Beim Schreiben ist es verständlicherweise nervig, wiederkehrende, lange Ausdrücke ausschreiben zu müssen.
Tipp: Reduzieren Sie Abkürzungen auf ein absolutes Minimum und verwenden Sie nur allgemein-gebräuchliche Abkürzungen. Das Lesen von Abkürzungen ist äusserst mühsam, insbesondere wenn eine Person diese nicht kennt. Oftmals wird der Aufwand beim Schreiben mit dem Aufwand beim Lesen gleichgesetzt, dies ist jedoch nicht der Fall. Beim Lesen überfliegt man die ausgeschriebenen Ausdrücke genauso schnell oder sogar noch schneller als die Abkürzungen. Im schlimmsten Fall kann es zu Missverständnissen kommen, wenn die gleiche Abkürzung mehrere Bedeutungen hat. Um den Schreibaufwand zu reduzieren, können Abkürzungen während des Schreibens verwendet und am Schluss mithilfe der Ersetzen-Funktion (Strg+H in MS Word) ersetzt werden. Man sollte jedoch auch an die Mitautor:innen denken, die möglicherweise viel Mühe darauf verwenden müssen, Abkürzungen zu entschlüsseln. Ein No-Go sind viele Abkürzungen in Protokollen, die durch externe Gutachter:innen beurteilt werden sollen. Wenn Abkürzungen verwendet werden, ist es unerlässlich, dass sie beim ersten Auftreten im Text (am besten pro Kapitel oder sogar Abschnitt) eingeführt werden und (!) ein vollständiges Abkürzungsverzeichnis erstellt wird (dieses sollte man bei jeder neu eingeführten Abkürzung sofort aktualisieren).
REDCap Light and user access rights to the database: What do I have to consider before I set my database into production status regarding role and access management?
Case
A monitor of a study still had data entry rights after a REDCap Light database was set to production status.
Considerations
Regarding the user access rights in REDCap Light databases please consider the following:
- Please check Chapter 6.1 and 7 in the REDCap Light Manual of CTU Bern regarding prerequisites for deployment and user/role management.
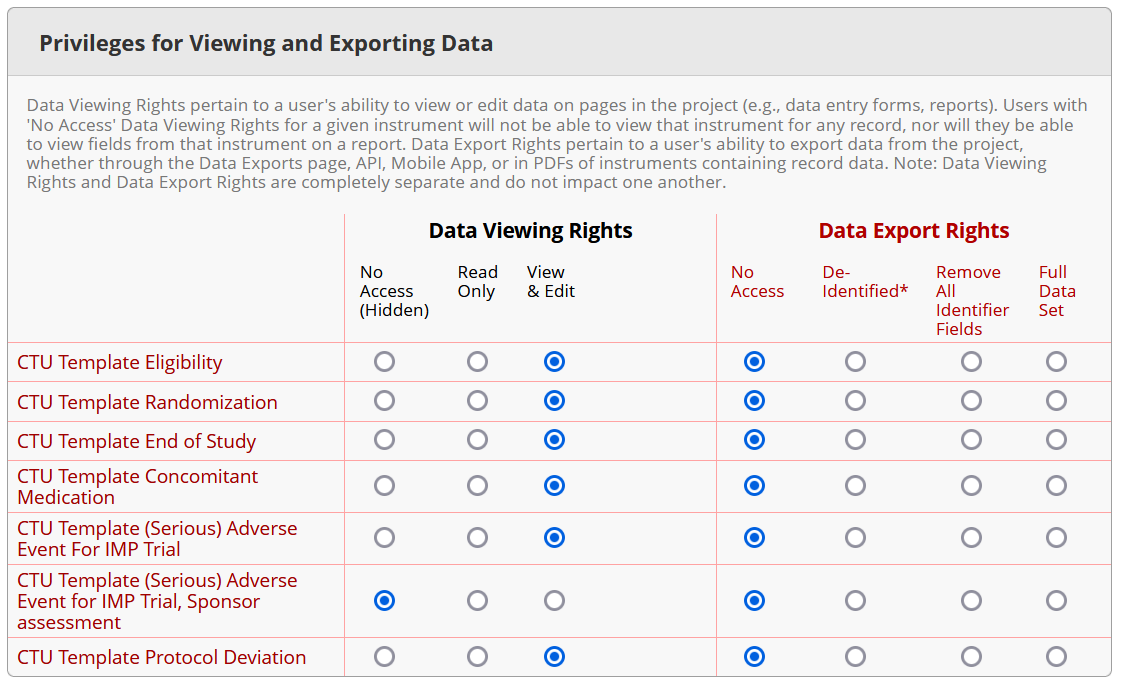
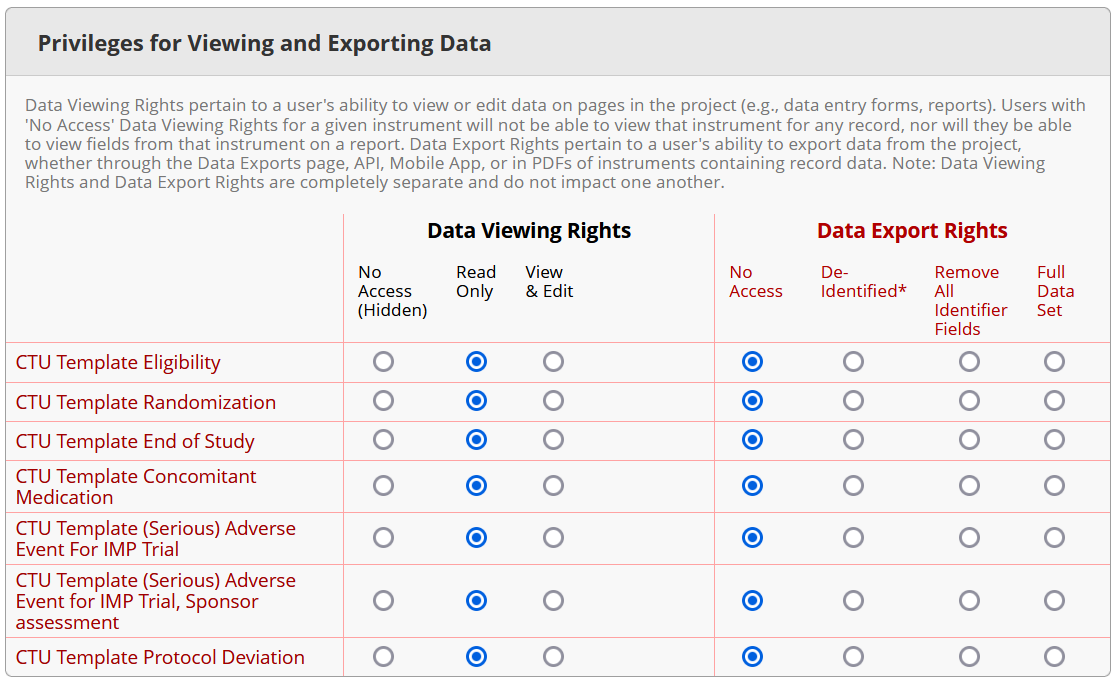
- IMPORTANT: Make sure that you have created user roles for all access types (e.g., data entry, monitoring, analysis…) or use the CTU Bern standard roles and verify that all role settings are accurate; also check the detail page of each role regarding data viewing rights and export rights.
Example for role ‘Data entry’
Example for role ‘Monitoring’
- Verify that all users who can access the project are assigned to a role. If there are users that are not assigned to a role, assign them to the correct role.
- Please note that CTU Bern Data Management must not assign users to a REDCap Light project and its roles. The REDCap Light project super user is responsible for this task. Also, CTU Bern staff (e.g., monitors, statisticians) need to be assigned by the project super user. CTU Bern Data Management only creates user accounts as requested by the sponsor/study team of the REDCap Light Service.
Lesson learned
BEFORE setting a database productive, it is important to check all user roles in detail under ‘User Rights’ in REDCap to verify that each role has the correct settings for data viewing and export rights.
Why am I usually not the sponsor of the trial that I initiated?
According to ICH-E6 (GCP), the sponsor is “an individual, company, institution, or organization which takes responsibility for the initiation, management, and/or financing of a clinical trial”.
GCP thus implies that indeed, a person can act as the sponsor of a clinical trial. Usually though, a natural person (physician in a hospital or employee of a pharma company) will not personally be able (and willing) to take on responsibility for a clinical trial with all its consequences (including e.g., liability). In most cases, the institution where the person is employed (hospital or company) will thus assume the title Sponsor. As an institution cannot act by itself, a (natural) person must represent the sponsor and act on its behalf while not being the sponsor him- or herself. In academia, this should usually be the person who had the idea for the project and initiated it. In industry, this is often the medical lead or medical officer. (Along those lines, we advise against using the term Sponsor-Investigator as a title, as this implies that a natural person assumes the role of the sponsor.)
In a clinical trial protocol, we thus recommend displaying the sponsor role as follows:
- Sponsor:
Wiesenspital
Birkenstrasse 20
5702 Frühlingsparadies
Represented by:
Prof. Dr. med. Vreni Pollenflug
Allergologische Klinik
Tel: 123 456 78 90
Email: vreni.pollenflug@wiesenspital
Nevertheless, for ease of everyday use, the person that initiated the trial, can call him- or herself “Sponsor”. It is just good that this person understands who the true sponsor of the study is.
For further reading on the proper use of the terms Sponsor, Sponsor-Investigator, and Investigator, see the CTU Newsletter from February 2023 below.
How can I safely change a question in a case report form (CRF) during data collection?
It is not unusual that a CRF needs to be adapted during the conduct of a study, e.g. following a protocol amendment or due to unclear wording of questions or missing choices. We covered the question of “what do I need to consider when changing a database during data collection?” in a previous lessons learned. Here we focus on the aspect of how to change an already existing question in one of your CRFs.
If data has already been collected, you might run into the major issue of corrupting data collection by changing a question without further considerations. We would like to illustrate this based on two examples:
Example 1: Adding choices to a radio button or a dropdown list
When adding a new choice it might be useful to rearrange the order. While changing the order is not an issue per se, never change the coding of your choices.
The original question with 4 choices (grey background) and the coding (white background):

The question with 2 choices added (Vaccination Center, Other):


Example 2: Changing the question text
Changing the wording of a question can have a serious impact on your already collected data.
The original question was formulated too broadly:

You want to amend the question and include a time period, in this case ‘during the surgical intervention’. Never change the original question, this will lead to a mismatch between the already collected data and the new wording of the question.
Rather add a sub-question that allows adding more specific information.

Lesson Learned:
A seemingly little change can have a big impact on your data collection! Therefore, when changing a CRF, always consider the potential influence on already collected data.
Furthermore: For both examples you need to decide how to handle the newly added possibilities for all previously entered data: will data be corrected/added retrospectively? And - don’t forget to inform all stakeholders concerned (e.g. participating study sites, on-site and central data monitors).
Was sind anonyme (Forschungs-)Daten?
Häufig taucht die Frage auf, ob ein bestimmter Datensatz aus anonymen Daten besteht (und damit jegliche Forschung nicht unter das Humanforschungsgesetz fällt). In diesem 9-minütigen Podcast erklären wir, was anonyme Daten sind und warum die CTU Bern davon ausgeht, dass im Bereich personenbasierte Gesundheitsforschung praktisch nie anonyme Daten vorkommen
Was sind verschlüsselte (Forschungs-)Daten?
Als Ergänzung zum Podcast zu anonymen (Forschungs-)Daten erklären wir in diesem 12-minütigen Podcast, was verschlüsselte Daten sind. Wie bei den anonymen Daten geht die CTU Bern auch bei verschlüsselten Daten davon aus, dass diese im Bereich personenbasierte Gesundheitsforschung praktisch nie vorkommen. Warum dies so ist und warum Forschungsdaten trotzdem pseudonymisiert werden müssen wird ebenfalls erklärt.
REDCap Light Services and user management: What do I have to consider when creating data access groups / sites for my trial in REDCap?
In a REDCap Light project, the project super user has the necessary access right to handle the user management on his own. This encompasses tasks like creating user roles with specific access and editing rights to the trial’s electronic case report forms, assigning users to these roles and creating data access groups (DAGs) for the sites involved in the trial.
Case: The randomization of a patient was not possible in the REDCap setup by the Principal Investigator; instead an error message occurred in the system. Upon review of the database through CTU Datamanagers it was identified, that the randomization list has had not appended to the database.
Considerations: When creating data access groups for a new site and assigning users to it, please consider the following:
- You need to assign a user to a specific site before they create the first record in the system, otherwise the record will not be visible for other members of the DAG. Furthermore, if you do not assign a user to a DAG, they will see all records entered in the study database.
- Super users themselves should never be allocated to a DAG! Otherwise, they will no longer be able to assign other users to DAGs. If a super user needs to create records and enter data, please request a second login which can then be used for data entry.
- IMPORTANT: If you have a randomization set up for your study and randomize by DAG/site, then you must append the uploaded allocation table by allocation results for the newly created DAG! Otherwise, it will not be possible to randomize patients since no randomization results are available for the new DAG.
Lession learned: Before adding new DAGs / sites to your study database and assigning users to them, you have to consider the dependencies and consequences for the study as a whole. Especially for randomized trials, make sure to append the randomization table before the first record of a newly added DAG is randomized!
What should I pay attention to when defining fields in REDCap?
The case: In a data exportation process from REDCap to an Excel file, problems occurred because fields intended to be numeric fields with two decimals, were set up as text fields in REDCap instead. As a result, multiple catastrophic errors occurred during the data export process from REDCap and subsequent import into Stata and R, which were haphazardly detected by comparing the number of missing values with the expected number of missing values.
What happened?
The fields contained strange numbers of the form xx.xxx.xxx due to numeric fields coded as text fields. The copy-paste action of data into these text fields lead to the errors (e.g. correction overwrite 51.11 with copy paste 49.2 leads to 49.251.11). Stata and R handled the import differently and returned wrong results, as both softwares tried to reformat the text field into a numeric field. The import as text only showed the mistakes.
Lesson learned?
It is a common mistake to define numeric fields as text fields within REDCap. However, this leads to substantial problems with statistical software (e.g. R, SPSS, Stata), as this software cannot readily analyze text fields and hence each software will make an estimate of what the text field should contain and may attempt to import it as a numeric field. You should always avoid using text fields except they really only serve to further describe e.g. a procedure or an adverse event. Instead, define numeric fields including defining the number of decimals needed.
What do I need to consider when changing a database during data collection?
It is not uncommon that a productive study database needs to be changed / adapted during data collection, for example due to a protocol amendment. When changing a productive database however, you need to carefully analyze the risks of the change.
Let us illustrate this on the example of the addition of a new eligibility criterion.
If CTU Bern is setting up the database for you, then the data management division will, for every major change, undertake a risk analysis. The goal of this is to identify and assess potential negative consequences of the change and define measures in order to mitigate the risks. Most importantly, CTU Bern makes sure that no data is lost and that the integrity of the database is ensured.
Let’s take following example: In a study, a new eligibility criterion is added during data collection. Following risks exist:
- Eligibility calculation works incorrectly for new or already recruited patients
- Records might be inconsistent since records that fall under the new protocol version but entered before the update of the database lack the entry for the new exclusion criterion
- Data might be lost if data fields are hidden based on the eligibility determination
CTU Bern then plans and documents the measures to minimize the identified risks. In this example, we create a test database where the change is implemented and then reviewed and tested by a second data manager. Only if all tests are successful, the change can then be implemented in the original database.
If you go for a REDCap Light Service and set up the database on your own, then it is in your responsibility to identify and assess the potential risks of a change. As a help, please refer to the REDCap Light User Manual section “9.2 Changes in a productive database”. Here you find important aspects to consider and tips when implementing changes during data collection.
=> When adding something new, this might negatively influence your already collected data. It is therefore of utmost importance to carefully evaluate the possible risks of any change to your database.
Mysterious lab sampling time points - What could be the explanation for this mystery?
In a recent trial that was monitored by CTU Bern, lab samples had to be taken within 10 Minutes prior and 2 hours after administration of study drug (i.e. a -10 minutes and a +2 hours sample), and lab results including sampling date and time point had to be recorded in the study database (the eCRF).
For 5 patients that had their visit early in the morning on the same day, it was noted by the monitor that no lab values were recorded in the eCRF for the +2 hour sample while the -10 minutes values were all there as needed.
The study nurse was convinced that she had taken all samples as required but assumed that the +2 hour samples must have been lost somewhere on their way to the central lab although she stated that this was quite unlikely.
As turned out later, she had indeed drawn all samples as required, they were all analysed by the central lab, and results were all available on the lab reports.
What could be the explanation for this mystery?
It’s actually pretty simple: The time points of the lab results that are indicated as “Probenentnahme” (sampling) on the lab reports are not the time point when the samples were drawn but when the tube labels were printed.
The tide of events was the following:
To prepare for the labour intensive early morning visit with the 5 patients, the study nurse had pre-printed the labels for the -10 Minutes samples in the evening before the visit. She then draw the samples at the right time point on the next day, attached the pre-printed labels (that had the date of the day before) to the tubes and sent them for analysis. She then also immediately printed the labels for the +2 hour sample. Two hours after drug administration, she attached the labels (with time point a few minutes before drug administration) on the +2 hour sample.
When completing the eCRF a couple of days later, she recorded the values of the +2 hour samples for the -10 minutes time point, because the lab report misleadingly indicated that sampling was done within the 10 minutes window prior to drug administration. And since on the lab reports, the results for the +2 hours were apparently missing, the corresponding fields in the eCRF were left empty.
Lessons learned?:
Be careful with lab sampling time points in clinical trials, in particular when lab sampling time points are important (and they are in many cases).
In the present case, the study nurse will now record the actual lab sampling time points together with the time point when the label was printed. Since only this will allow her to unambiguously link the results on the lab reports to the samples she actually drew. She will also prepare a note to file to explain that the “Probenentnahme” time point on the lab report is actually the time point when the tube label was printed.
Who should get a GCP certificate in a study team?
Short answer: Knowledge and compliance with GCP is necessary for anyone involved in a clinical trial.
Long answer: The Swiss law require that a clinical trial investigator should be qualified by education, training and professional expertise to assume responsibility to conduct a clinical trial, with adequate training in Good Clinical Practice explicitly being mentioned (ClinO, Art 6. Para.1).
Based on these legal requirements, swissethics defines a GCP Course on Investigator Level (“Basic GCP”) as mandatory for investigators in clinical research (swissethics.ch/en/aus-fortbildung > For investigators).
Furthermore, according again to the “Clinical Trials Ordinance (ClinO)”, other persons involved in a clinical trial must have the professional knowledge and experience appropriate to the activities in question (ClinO, Art 6. Para.4). Therefore, holding a GCP certificate it’s not a legal requirement for persons other than an investigator involved in a trial.
However, the CTU Bern (as well as Swissethics) strongly recommends training in relevant aspects of GCP for anyone involved in a clinical trial.
Although neither the Swiss law nor GCP mentions mandatory education requirements for a study sponsor, Swissethics requires training in research ethics and GCP on a Sponsor-Investigator level (Advanced GCP) for Sponsor-Investigators, e.g. Investigators who both initiate and conduct, alone or with others, a clinical trial.
To certify the training, involved personnel should hold a valid and recognized GCP certificate (a list of recognized GCP course providers is available on the website of Swissethics > Aus-, Fortbildung). Alternatively to GCP courses, a recognized certificate can be obtained after completion of an online course (http://elearning.trree.org/). The frequency of GCP re-training is not defined in the regulations, nor in the Swissethics guideline, but it’s highly recommended to participate to a GCP refresher course every once in a while. Note that CTU Bern is offering a GCP Refresher course annually. Check the website for dates or subscribe the CTU Bern newsletter to be informed about current course dates.
What does the Mann-Whitney test actually test?
The Wilcoxon rank-sum test which is also known as Mann-Whitney U test or Mann–Whitney–Wilcoxon test is arguable the most famous and frequently used non-parametric test to compare two groups. Very often, only a p-value is given as a result even though it is good practice and generally recommended to report a measure of effect size for an inferential test. The reason may be a widespread misconception about what the test actually tests and whether a usable measure of effect size is available.
It is a widely believed that the Wilcoxon rank-sum test is a test for equality of medians, which is only true under very specific circumstances—i.e. under the strong assumption that both of the two distributions are symmetrical about their respective medians or that the distributions are of the same shape but differ in location. In many situations, the median difference is therefore not a good measure of effect size.
What the test actually does was stated by Mann and Whitney in the title of their famous paper on the extension of Wilcoxon’s test: “On a test whether one of two random variables is stochastically larger than the other”. In other words, it tests whether an observation from one group is larger than an observation from the other group, or—in the context of a clinical trial—whether a patient in the treatment group has the better outcome than one in the control group.
The corresponding measure of effect size is the probability that a patient in the treatment group will have a better outcome than a patient in the control group—a quantity of practical interest that does not depend on the unit of the outcome. We suggest reporting this “Mann-Whitney statistic” with a confidence interval together with the p-value from the test.
Unfortunately, the Mann-Whitney statistic goes under various names—e.g. probabilistic index, measure of stochastic superiority, or common language effect size—and it is not always reported by statistical software packages. As a workaround, the well-known area under the receiving-operator curve (AUC or just C-statistic) can be reported—it is nothing else than the Mann-Whitney statistic.
I don’t have final approval from the ethic commission; can I start to register my study at clinicaltrials.gov and kofam.ch (SNCTP)?
According to Swiss law, a clinical the trial must be registered on the Swiss Clinical Trials Portal SNTCP (kofam.ch) before the official start of the trial. Moreover, the trial must be registered in a register that meets WHO registry criteria such as for example clinicaltrials.gov.
When filling out the application for the ethic commission (BASEC), it is recommended to agree to the automatic transmission of the trial’s info to the SNTCP portal. The info concerning the trial will be automatically transferred to the portal as soon as the trial is authorized and an external identification number is available (e.g. NCT12345678). This number is automatically generated by clinicaltrials.gov as soon as you start registering a trial.
It is possible to start adding info about the trial on clinicaltrials.gov at the time of your initial submission to the ethics commission but you may not yet have all the information to fill in the required fields. If you do so, please set the study to: “Not yet recruiting”. It is possible and actually advised to start and then add the information later, after receiving formal approval from the Ethics Committee.
Make sure that the information on kofam.ch and clinicaltrials.gov match the trial protocol and are up to date before including patients.
Can I let study participants sign the Informed Consent Form electronically on the Electronic Data Capture System (EDCS)?
To be recognized as legally valid, or in other words, to be considered as fully equivalent to handwritten signatures, electronic signatures must comply with the requirements of a «qualified electronic signature» as stated in the Federal Code of Obligations (Art. 14 Abs. 2bis)
The “Federal Law regarding the electronic signature” regulates and defines the requirements for the qualified electronic signature:
- it is allocated exclusively to its owner,
- it enables the identification of the owner,
- it is created by means which are exclusively controllable by the owner herself/himself
- the link between the electronic signature and the data to which it refers, is implemented in a way that allows for the detection of subsequent modification of said data» (Swiss Federal Law regarding the electronic signature, Art. 2b, https://www.admin.ch/opc/de/classified-compilation/20131913/index.html)
In addition to the specifications regarding the signature itself, the Swiss Federal Law also stipulates how the creation process of the signature must be executed. The generation of electronic signatures must adhere to a specific authentication process, which can only be carried out by providers who have been validated by the federal administration.
In Switzerland, solely the „SuisseID“complies with all legal requirements as outlined above. It is offered for example by the Swiss Post, who is recognized as an accredited provider
The electronic signatures, which can be generated by the EDCSs used in clinical trials, do not comply with the legal requirements regarding validated electronic signatures effective in Switzerland. Consequently, the EDCS-generated signatures cannot be used on the Informed Consent Form, whether for the signature of the study participant nor for the signature by the Principal Investigator.
Can I send the randomization list to the principal investigator in an open-label study?
No it is not - as a matter of fact, it is highly problematic. One of the key features of an RCT is the concealment of the allocation sequence: the person who is responsible for the inclusion of participants should have no knowledge about the type of intervention allocated to the next participant. This knowledge might influence the decision of the enrolling physician whether or not to include a specific prospective participant. In short, knowledge about the allocation sequence could lead to selection bias in RCTs. Consequently, it is essential that the randomization list is password-protected and concealed from study personnel involved in patient recruitment at all times. Only the collaborators that generate and implement the list (i.e. statistician, data manager) should have access.
Is it acceptable to appoint more than one sponsor to a clinical trial?
4 investigators in the UK, Germany, USA, and Switzerland plan to perform a drug trial with participating sites in all four countries, and all four investigators are named as co-sponsors in the protocol. However, according to the “AW-Information Sheet FAQ on clinical trials with medicinal products”, Swissmedic accepts only one sponsor for a clinical trial who must assume the overall responsibility for the clinical trial in Switzerland. How should this be handled?
To ensure that the responsibilities are clearly defined, only one investigator (or the institution he or she is associated with) in either one of the four countries should be appointed to the role of the sole sponsor, i.e. taking over the overall responsibility for study initiation and conduct and this should be displayed in the protocol accordingly.
In the other countries sponsor-representatives must be installed. If two or more countries are part of the EU, for those countries only one sponsor-representative in either one of those countries is required.
The responsibility of a sponsor-representative is usually taken over by a contract research organization, a country affiliate of the sponsor (if the sponsor is e.g. a pharma company with headquarters abroad) or another institution located in Switzerland and specialized in this field of work. It is recommended not to delegate this function to the individual investigator, since in the worst case, he or she might be personally confronted with liability and coverage claims, which may be difficult for the investigator to cope with. For studies conducted at Insel, an application can be made to the Departement für Lehre und Forschung (DLF) so the investigator can take over this function without taking personal risks.
In Switzerland the sponsor-representative takes over the role of the communication partner with the authorities during the approval process as well as other responsibilities, which are described in the Interpretation guide – Obligations of representatives of foreign sponsors, version 1.1 (link see below) from the Federal office of public health (FOPH).
Note that for non-drug / non-medical devices trials, co-sponsorship may be acceptable but this needs to be discussed with the responsible local authorities on a case by case basis since such trials may not be regulated in foreign countries.
Can I exclude a randomized patient from a study?
A new device is compared against the standard device in a randomized controlled trial (RCT). A patient is randomized to the new device before the surgery. During the surgery, complications occur. Therefore, it is decided to use the standard device because the operation team feels more familiar with that device. Since the patient did not receive the randomized device, no data was collected and no further follow-up was performed for this patient. Is this correct?
No, this is not correct. A randomized patient may never be excluded from a study in retrospect. Every effort has to be taken to collect all baseline data, all procedural data, and all follow-up data, also of patients that did not receive the correct intervention or patients with major protocol violations. Only then, an intention-to-treat (ITT) analysis is possible, which is the gold standard in RCTs. This means that all randomized patients are evaluated in the group they were originally allocated to, regardless of the intervention actually received.
When can I collect identifying data and what shall I do if I participate in a study that is collecting identifying data?
The case: CTU Bern had been asked to set-up a study database including electronic case report forms (eCRF) for a (national) multicenter RCT. The investigator asked whether it would be possible to also store names and contact details of participants within the database. She argued that this would make follow-up phone calls much easier. Based on a detailed assessment of the relevant regulations (Human Research Act, Human Research Ordinance), information from ethics committees, and personal discussions with the cantonal ethics committee the decision was taken not to store this identifying information centrally but rather only locally (paper or Excel code list, depending on the site).
Lessons learned: Generally, a research database should not contain any identifying data. Examples of identifying data are: names, date of birth, addresses including phone numbers, but also: initials, patient identification numbers (e.g. Insel PID), case identification numbers (e.g. Insel FID), and e-mail addresses. Not identifying data according to swissethics are: Year of birth and gender.
If you are participating in an international study that is collecting identifying data, you should enter dummy information for the fields that are not allowed in Switzerland (e.g. ‘A.A.’ for the initials or ‘01.01.YYYY’ for the date and month of birth). Also refer to the guideline on coding of trial subjects accepted by swissethics which can be found in the template section of swissethics website (http://www.swissethics.ch).
In special cases the collection of identifying data might be needed (e.g. addresses for follow-up visits if these cannot be stored otherwise e.g. if reminders/forms are send by e-mail, month or even date of birth for studies with children). In this case, you must specify in the study protocol as well as in the informed consent that identifying data are being collected and you must add a justification why it is needed. The ethics committee will decide if they agree to the collection of identifying data.
Even if you are allowed to collect identifying data you should make sure that you protect them well, i.e. that you restrict access to this data to the people who need it e.g. addresses should only be accessible to persons who actually need to contact the patients.
What is a Site Delegation & Signature Log and what purpose does it serve?
A site delegation & signature log (also called authorization log, authorized signature log, or site delegation log) is an important document that should be implemented in all clinical trials. On this log, the principal investigator lists study staff who is authorized to participate in the clinical trial and documents which task each person is allowed to perform. Its aim is also to give a sample of the signatures and (hand-written) initials of study staff so it can be reconstructed which staff member signed or initialed a study document. This document should not be confused with the List of Study Staff submitted to the EC during the protocol submission process or other similar lists, since these may not fulfill the requirements of a site delegation log. A template can be downloaded here.
I forgot to (sign and) date an informed consent form / case report form when I completed it. Can I simply add the date of completion & sign it?
No, backdating is never allowed. You should always add the current date, ideally with a comment “signed for [date when you originally completed the document]” (e.g. “Lieschen Müller, 03.10.2016, signed for 02.03.2016”). The reason for this is that one should be able to reconstruct when documents were completed & signed & when later entries (in this case e.g. the signature) were added. If you simply backdate documents, it looks as if you dated the document at the time of completion, which is not the case.